Colored Design
Welcome to the web site on disordered systems
This page has been designed to be an internet source that presents theoretical modelling of complex and disordered materials such as glasses, amorphous systems, liquids, granular materials, etc.
Rigidity and topological phases of amorphous networks
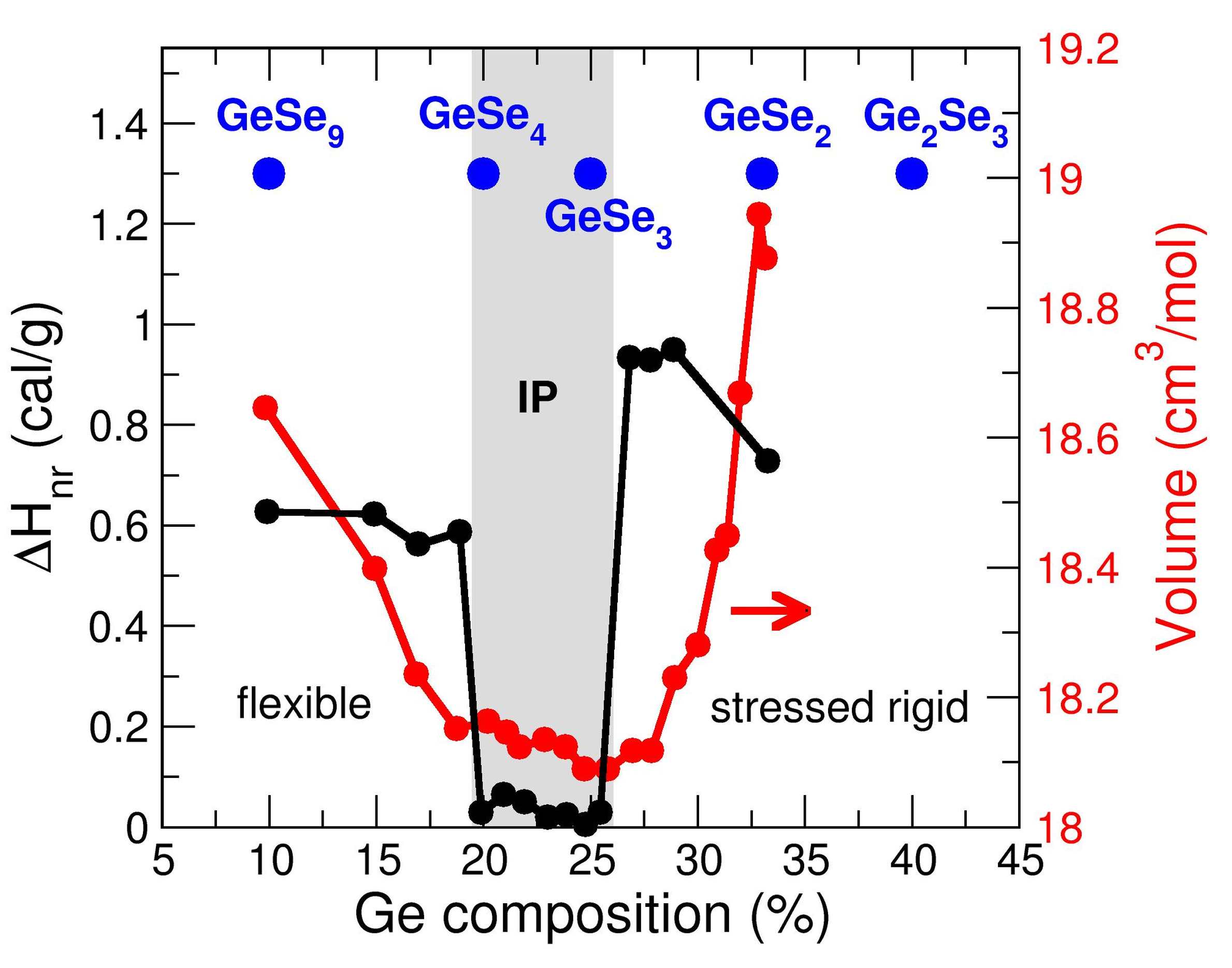
What happens if the atomic bond density changes in an amorphous network ? A mean-field treatment at zero temperature using Maxwell theory of elasticity has predicted a flexible to rigid transition, located at the Maxwell isostatic stability condition. Experimentally, there is now growing evidence that the locus of the transition is not unique and that glassy relaxation effects are present. This forces to reconsider concepts and results from the initial rigidity theory, and has led to the identification of a new topological (intermediate) phase with anomalous properties that are now fairly well characterized. See :
- Densified network glasses and liquids with thermodynamically reversible and structurally adaptive behavior, M. Bauchy, M. Micoulaut, Nature Communications 6, 6398 (2015)
Ab initio and classical simulations of liquids and glasses
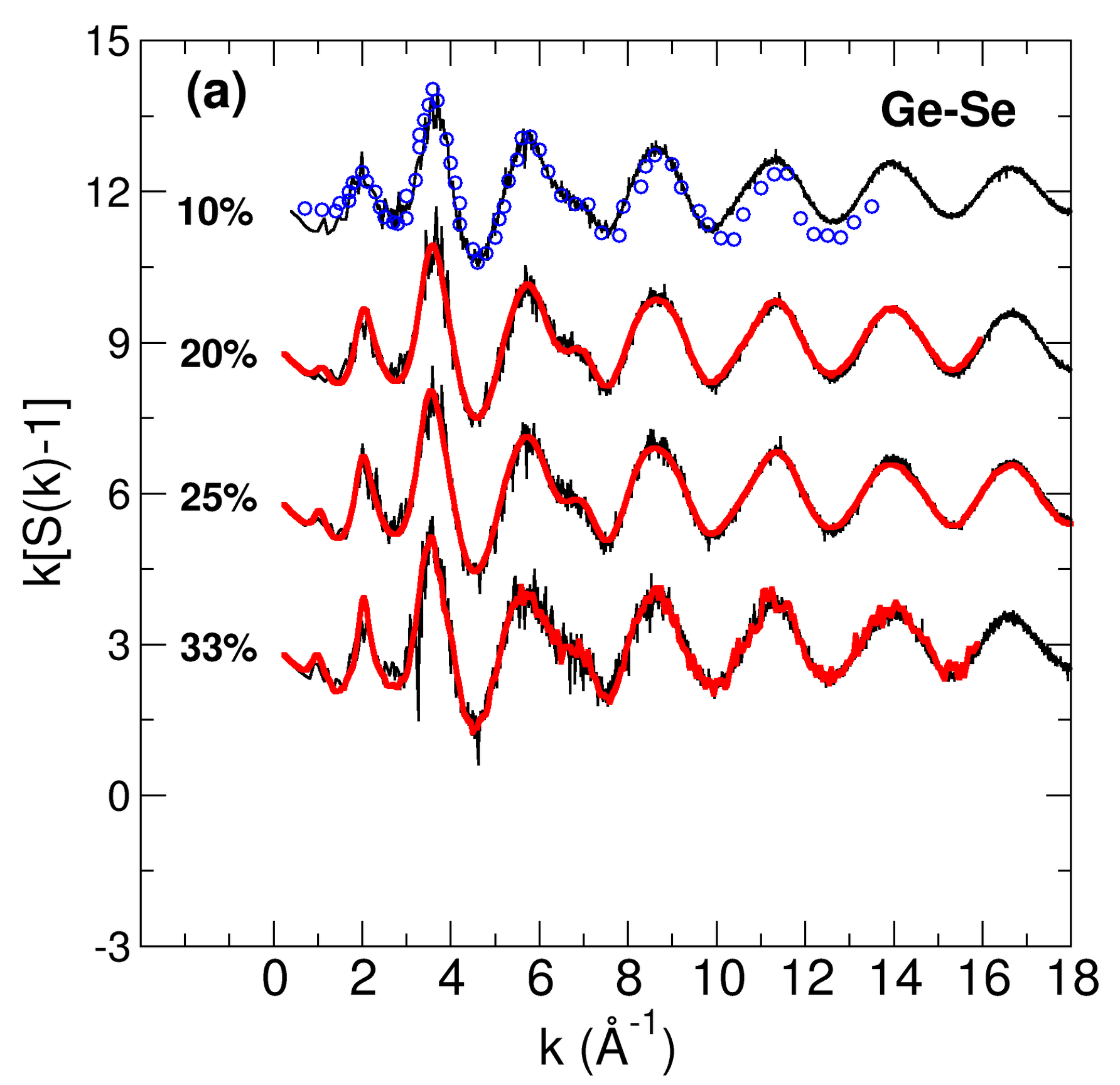
Molecular simulations are very helpful in providing a detailed atomic scale description of various properties for a variety of materials. Depending on the nature of the chemical bond, one uses either a classical force-field (for ionic systems) or a quantum treatment in order to treat correctly the charge transfer with time (for covalent systems).
We have spent time and efforts in improving the description of archetypal liquid and glassy chalcogenides
(GeSe2, As2Se
- Crucial Role of S8-Rings in Structural, Relaxation, Vibrational and Electronic Properties of Liquid Sulfur close to the lambda Transition, H. Flores-Ruiz, M. Micoulaut, Journal of Chemical Physics 157, 054507 (2022)
Fast ion conductors

The worldwide ubiquitous demands for energy in vehicles and portable electronics have exploded the demand for higher power density batteries. In this perspective, amorphous electrolytes have been identified as a promising path for the next generation "all solid state" batteries. There is, however, need to improve our understanding of the effect of atomic scale properties which influence the ion conduction in a disordered media or to establish the role of the migrating ions.
We have recently investigated certain sulfide glasses using phenomenological models and molecular simulations, able to describe electric properties in connection with other important structural features. See :
- Fast ion conduction and flexibility and rigidity of solid electrolyte glasses, M. Micoulaut, M. Malki, D.I. Novita, P. Boolchand, Physical Review B 80, 184205 (2009).
Polyamorphism
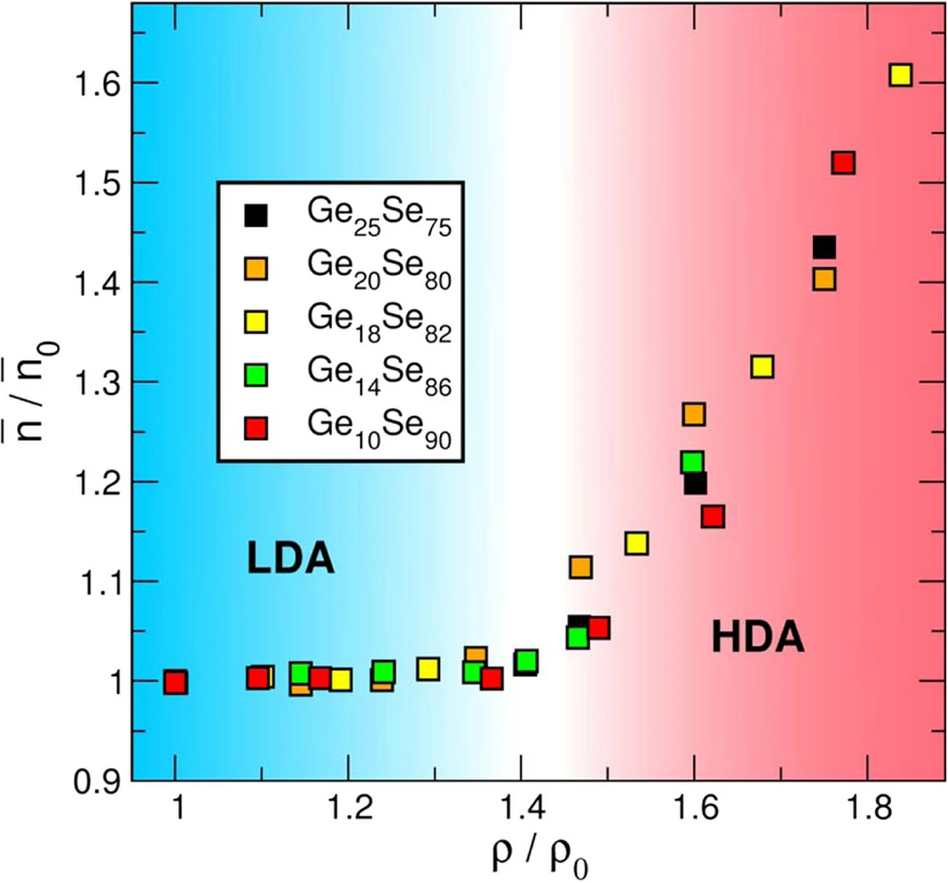
Pressure-induced phase transformations have attracted widespread interest in condensed matter science. Analogous polymorphism observed in crystals, the observation of metastable configurations in the pressure an amorphous system has led to the identifcation of polyamorphic transitions coupled to liquid-liquid transitions driven by coordination changes with densification.
In combination with experiments, we have characterized such transitions using ab initio molecular simulations for several important glassy systems : B2O3 and Ge-Se. See :
- Universal amorphous-amorphous transition in GexSe1-x glasses under pressure, C. Yildirim, M. Micoulaut, P. Boolchand, I. Kantor, O. Mathon, J.-P. Gaspard, T. Irifune, J.-Y. Raty, Scientific Reports 6, 27317 (2016)
Data storage at the atomic scale
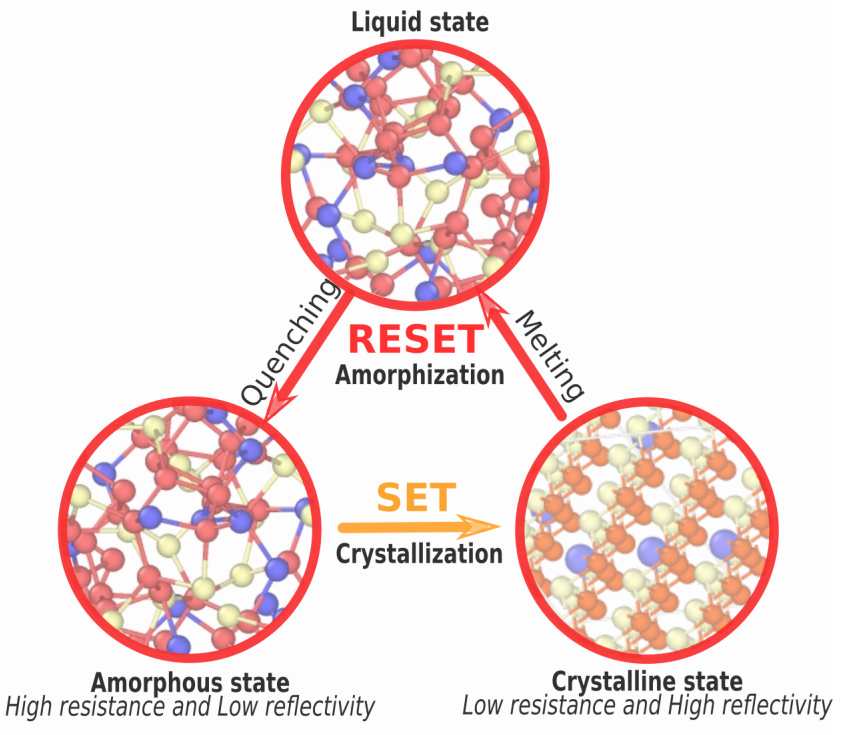
Chalcogenide based crystalline and amorphous materials can be used for a rather wide range of promising applications such as thermoelectrics, flash memory/PRAM devices or optical disk memories using the phase change mechanism, i.e. the rapid switching between a crystal and an amorphous phase. In all applications, the huge difference in electronic and optical properties between the crystalline and amorphous state is used. The origin of such differences is not fully understood which turns out to be driven by subtle changes in bonding properties altered by chemical alloying.
Using ab initio simulations, we have been continously improving the structural description of prototypal PCMs such as GeTe or Ge2Sb2Te5, and have shown that Ge atoms play a crucial role in the switching by having a predominantly tetrahedral ordering that impacts electronic properties close to the Fermi level. See:
- Alteration of Structure, Electronic, and Vibrational Properties of amorphous GeTe by Selenium substitution: an Experimentally Constrained Density Functional Study, M. Micoulaut, A. Piarristeguy, O. Masson, R. Escalier, H. Flores-Ruiz, A. Pradel, Physical Review B 104 (2021) 144204
Glass transition
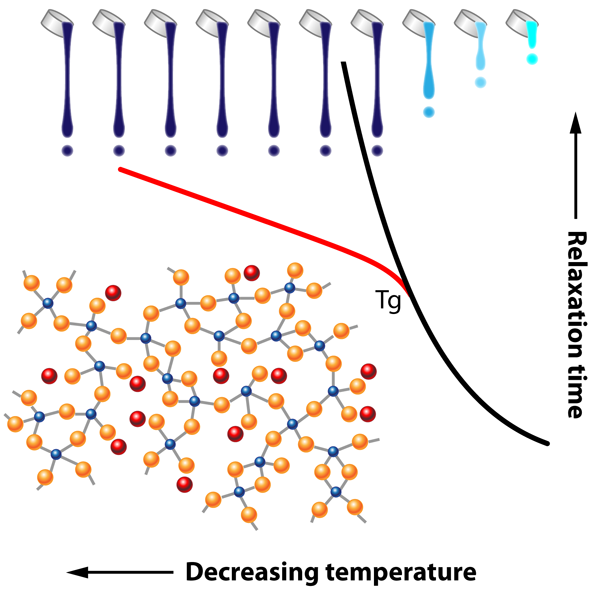
If crystallization can be avoided during cooling, a liquid will display a substantial increase of its viscosity, and will form a glass that behaves as a solid with a relaxation time that grows exponentially with decreasing temperature. Given this off-equilibrium nature, physical quantities now depend on the waiting time before a measurement is performed.
We have been using phenomenological models or molecular simulations to investigate the relaxation properties (dynamics, heterogenetities, Tg cycle,...) of variety of glass-forming liquids ranging from Lennardjonesium to more realistic systems including silica, GeSe2 or GST225. See:
- Relaxation and physical ageing in network glasses : a review, M. Micoulaut, Report on Progress in Physics 79, 066504 (2016)